We are looking for an experienced software engineer to join our full-stack engineering team. The team is responsible for developing cloud infrastructure to empower our internal software and algorithms in the drug discovery field. The team develops solutions ranging from web front-ends for internal algorithms and software products, to database systems managing our experimental projects and data, and complex distributed computation and job scheduling systems.
Please send your CV and cover letter to career@accutarbio.com.
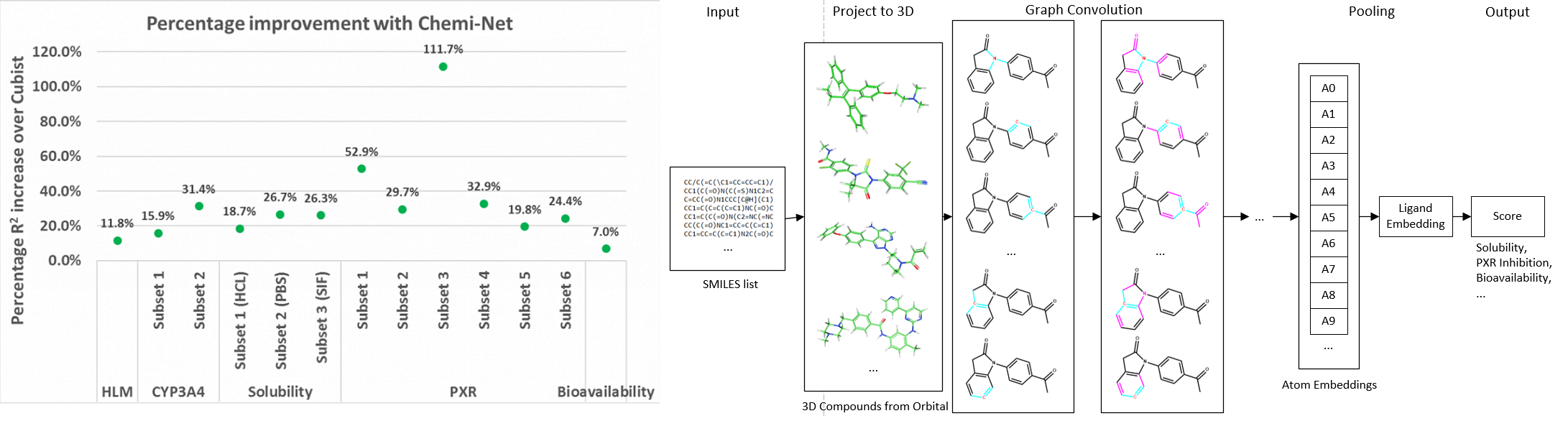
Figure. Left panel: Percentage R2 improvement over Cubist using Chemi-Net. Right panel: Overall network architecture.
Absorption, distribution, metabolism, and excretion (ADME) studies are critical for drug discovery. Conventionally, these tasks, together with other chemical property predictions, rely on domain-specific feature descriptors, or fingerprints. Following the recent success of neural networks, Accutar Biotech developed Chemi-Net, a completely data-driven, domain knowledge-free, deep learning method for ADME property prediction. To compare the relative performance of Chemi-Net with Cubist, one of the popular machine-learning programs used by Amgen, a large-scale ADME property prediction study was performed on-site at Amgen. The results showed that our deep neural network method improved current methods by a large margin. Accutar Biotech foresee that the significantly increased accuracy of ADME prediction seen with Chemi-Net over Cubist will greatly accelerate drug discovery.
We are looking for a talented individual to join our algorithm team to solve some of the most challenging algorithm problems in the world.
Our algorithm team is responsible for developing state of the art algorithms to implement our drug discovery methodology. We work with our biology teams to develop novel models with a strong biochemistry background. We utilize algorithms in various areas including, but not limited to, computational geometry, machine learning, optimization theory, combinatorial optimization, and bioinformatics.
We not only focus on the performance of our algorithms, but also work closely with our product team. Our products are used by the world’s best pharmaceutical companies and research institutions. To accomplish this, we spend a lot of time making our algorithm run efficiently on major hardware platforms. Besides tuning the algorithm, we also develop custom distributed platforms, machine-learning toolkits, and 3D visualization tools to get the most from the hardware we use.
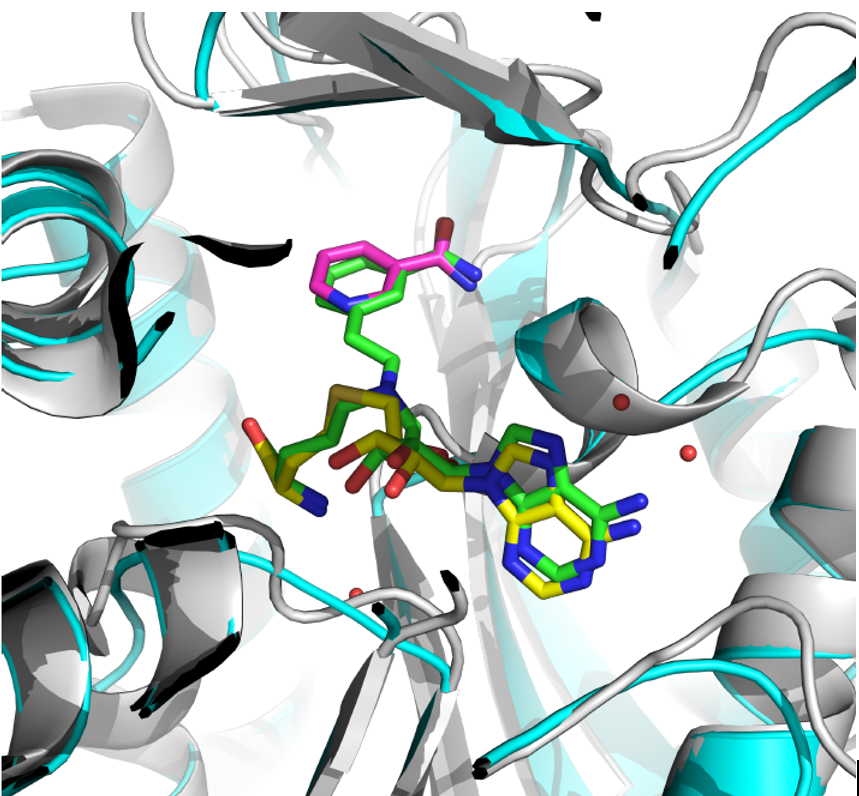
Figure: Side chain flexible docking model (in green) enables the chimera design of two ligand fragments (in yellow and pink).
A bisubstrate nicotinamide N-methyltransferase (NNMT) inhibitor was developed with the aid of Accutar Biotech’s platform recently. The binding pose of the designed inhibitor with human recombinant NNMT was predicted by Accutar Biotech’s side chain flexible docking method. The docking result was further validated by crystallization experiments.
A deep neural network based architecture was constructed to predict amino acid side chain conformation with unprecedented accuracy. Amino acid side chain conformation prediction is essential for protein homology modeling and protein design. Current widely-adopted methods use physics-based energy functions to evaluate side chain conformation. Here, using a deep neural network architecture without physics-based assumptions, we have demonstrated that side chain conformation prediction accuracy can be improved by more than 25%, especially for aromatic residues compared with current standard methods. More strikingly, the prediction method presented here is robust enough to identify individual conformational outliers from high resolution structures in a protein data bank without providing its structural factors. We envisage that our amino acid side chain predictor could be used as a quality-check step for future protein structure model validation and many other potential applications such as side chain assignment in Cryo-electron microscopy, crystallography model auto-building, protein folding and small molecule ligand docking.

Using a deep neural network architecture without physics-based assumptions, Accutar Biotech has demonstrated that side chain conformation prediction accuracy can be improved by more than 25%, especially for aromatic residues compared with current standard methods.