Prostate cancer (PCa) is primarily driven by aberrant Androgen Receptor (AR) signaling. Although there has been substantial advancement in antiandrogen therapies, resistance to these treatments remains a significant obstacle, often marked by continuous or enhanced AR signaling in resistant tumors. While the dysregulation of the ubiquitination-based protein degradation process is instrumental in the accumulation of oncogenic proteins, including AR, the molecular mechanism of ubiquitination-driven AR degradation remains largely undefined. We identified UBE2J1 as the critical E2 ubiquitin-conjugating enzyme responsible for guiding AR ubiquitination and eventual degradation. The absence of UBE2J1, found in 5–15% of PCa patients, results in disrupted AR ubiquitination and degradation. This disruption leads to an accumulation of AR proteins, promoting resistance to antiandrogen treatments. By employing a ubiquitination-based AR degrader to adeptly restore AR ubiquitination, we reestablished AR degradation and inhibited the proliferation of antiandrogen-resistant PCa tumors. These findings underscore the fundamental role of UBE2J1 in AR degradation and illuminate an uncharted mechanism through which PCa maintains heightened AR protein levels, fostering resistance to antiandrogen therapies.

We have developed an end-to-end, retrosynthesis system, named ChemiRise, that can propose complete retrosynthesis routes for organic compounds rapidly and reliably. The system was trained on a processed patent database of over 3 million organic reactions. Experimental reactions were atom-mapped, clustered, and extracted into reaction templates. We then trained a graph convolutional neural network-based one-step reaction proposer using template embeddings and developed a guiding algorithm on the directed acyclic graph (DAG) of chemical compounds to find the best candidate to explore. The atom-mapping algorithm and the one-step reaction proposer were benchmarked against previous studies and showed better results. The final product was demonstrated by retrosynthesis routes reviewed and rated by human experts, showing satisfying functionality and a potential productivity boost in real-life use cases.
We propose a method based on neural networks to accurately predict hydration sites in proteins. In our approach, high-quality data of protein structures are used to parametrize our neural network model, which is a differentiable score function that can evaluate an arbitrary position in 3D structures on proteins and predict the nearest water molecule that is not present. The score function is further integrated into our water placement algorithm to generate explicit hydration sites. In experiments on the OppA protein dataset used in previous studies and our selection of protein structures, our method achieves the highest model quality in terms of F1 score, compared to several previous studies.
Molecular modeling is an important topic in drug discovery. Decades of research have led to the development of high quality scalable molecular force fields.
In this paper, we show that neural networks can be used to train a universal approximator for energy potential functions.
By incorporating a fully automated training process we have been able to train smooth, differentiable, and predictive potential functions on large-scale crystal structures. A variety of tests have also been performed to show the superiority and versatility of the machine-learned model.
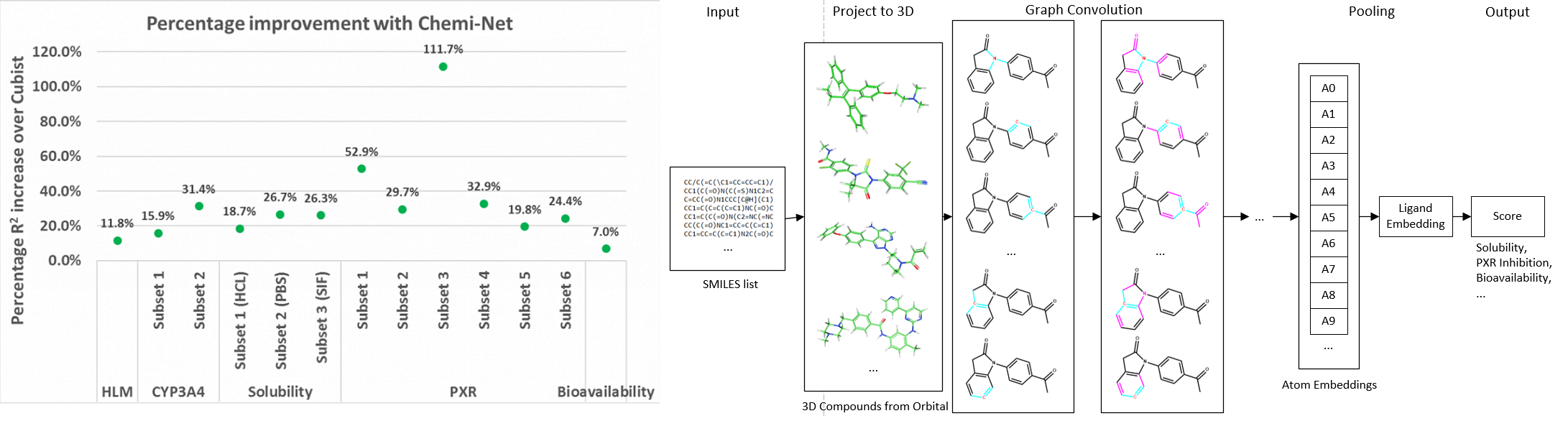 Figure. Left panel: Percentage R2 improvement over Cubist using Chemi-Net. Right panel: Overall network architecture.
Absorption, distribution, metabolism, and excretion (ADME) studies are critical for drug discovery. Conventionally, these tasks, together with other chemical property predictions, rely on domain-specific feature descriptors, or fingerprints. Following the recent success of neural networks, Accutar Biotech developed Chemi-Net, a completely data-driven, domain knowledge-free, deep learning method for ADME property prediction. To compare the relative performance of Chemi-Net with Cubist, one of the popular machine-learning programs used by Amgen, a large-scale ADME property prediction study was performed on-site at Amgen. The results showed that our deep neural network method improved current methods by a large margin. Accutar Biotech foresee that the significantly increased accuracy of ADME prediction seen with Chemi-Net over Cubist will greatly accelerate drug discovery.
Figure. Left panel: Percentage R2 improvement over Cubist using Chemi-Net. Right panel: Overall network architecture.
Absorption, distribution, metabolism, and excretion (ADME) studies are critical for drug discovery. Conventionally, these tasks, together with other chemical property predictions, rely on domain-specific feature descriptors, or fingerprints. Following the recent success of neural networks, Accutar Biotech developed Chemi-Net, a completely data-driven, domain knowledge-free, deep learning method for ADME property prediction. To compare the relative performance of Chemi-Net with Cubist, one of the popular machine-learning programs used by Amgen, a large-scale ADME property prediction study was performed on-site at Amgen. The results showed that our deep neural network method improved current methods by a large margin. Accutar Biotech foresee that the significantly increased accuracy of ADME prediction seen with Chemi-Net over Cubist will greatly accelerate drug discovery.